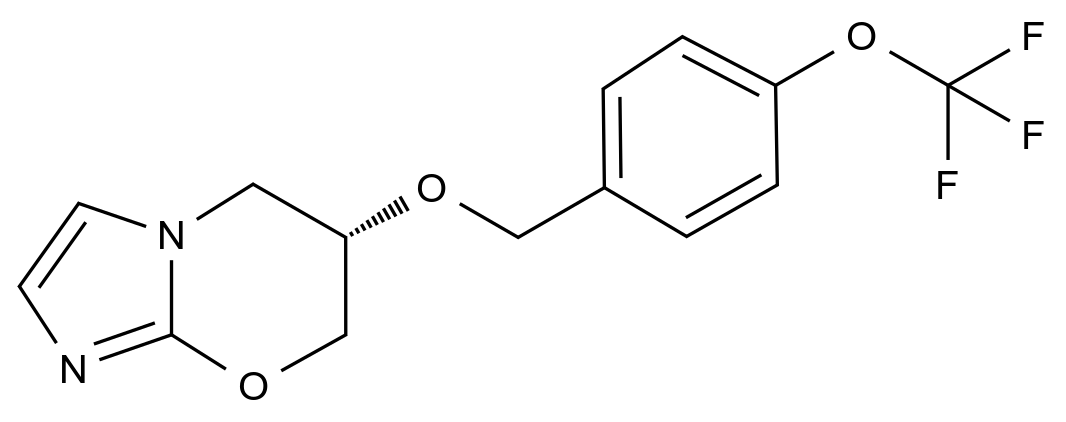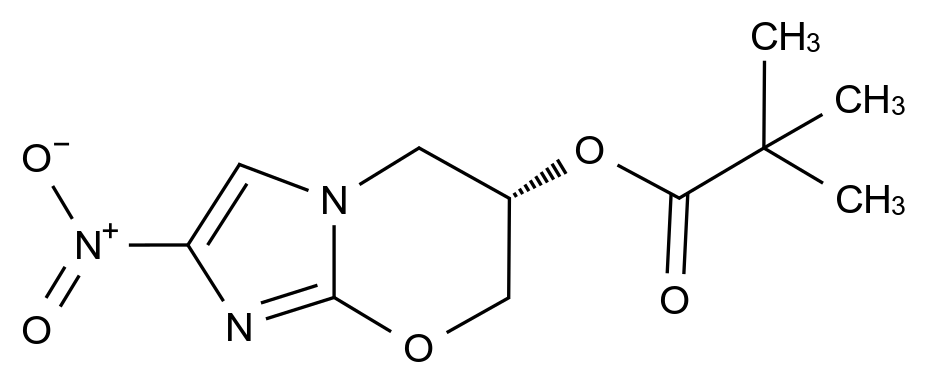
药品的质量控制是确保其安全性、有效性和一致性的关键环节。随着全球医药行业的不断发展,药品杂质的检测成为药品生产和监管中不可忽视的一部分。为了规范杂质的控制,世界范围内出现了多个药典,其中美国药典(USP)和欧洲药典(EP)是最为广泛使用的两大标准。USP与EP在药品杂质检测中所发挥的标准化作用,不仅为药品的生产企业提供了明确的质量控制指导,也为药品监管机构制定检测标准和实施法规提供了基础。
1. USP与EP的背景与定义
美国药典(USP):USP是由美国药典委员会(United States Pharmacopeia Convention)制定的药品质量标准,旨在为美国及全球的药品提供统一的质量、纯度和剂量标准。USP中的标准涵盖了药品的制备、检测方法及杂质控制。
欧洲药典(EP):EP由欧洲药典委员会(European Pharmacopoeia Commission)发布,是欧洲药品监管的核心标准之一。它为药品的生产和质量控制提供了具体的检测方法和要求。EP的标准同样涵盖了药品的所有方面,包括药品成分、纯度及杂质控制。
2. USP与EP在药品杂质检测中的作用
杂质检测主要针对药品中可能存在的有害物质,这些物质可能来源于原料、生产过程或存储条件。USP与EP分别针对药品杂质制定了详细的检测方法和控制标准,在药品的生产、质量保证和监管过程中起到了至关重要的作用。
2.1 标准化检测方法
USP与EP通过对药品杂质的检测方法进行标准化,确保了全球药品质量的统一。两者均制定了针对常见药品杂质(如溶剂残留、重金属、氧化产物等)的检测方法。例如,USP与EP均提供了针对亚硝胺、溶剂残留及微生物污染等的具体检测方法和检测限,这些方法包括高效液相色谱(HPLC)、气相色谱(GC)、质谱(MS)等技术的应用。
通过这些标准化的检测方法,药品生产企业能够一致地对其产品中的杂质进行检测,从而确保产品的质量符合全球监管要求。
2.2 控制杂质限度
USP与EP在药品杂质限度的设定上也发挥了重要作用。通过明确的杂质限度标准,药品生产商能够有效地控制药品中的杂质水平,从而减少对患者的潜在风险。例如,针对药品中溶剂残留的限度,USP和EP都设定了严格的限值,超出限值的药品将无法通过质量控制,无法上市销售。
USP与EP提供的标准不仅在数量上为药品杂质设定了限度,还在性质上定义了允许的杂质种类。通过这些限度标准的实施,全球药品的杂质控制趋于一致,减少了不同地区药品质量差异所带来的监管难度。
2.3 提供全球一致性
USP与EP的标准为全球药品质量提供了一致性基础。随着全球化药品市场的不断扩展,药品在不同地区的生产、销售和使用需要统一的质量标准。USP与EP为药品生产商提供了清晰、统一的杂质控制标准,帮助其产品满足全球不同市场的要求。尤其是对于跨国药企,遵守USP和EP的标准可以减少因杂质问题而引发的合规风险和法律纠纷。
3. USP与EP的差异
尽管USP和EP都致力于为全球药品提供统一的质量标准,但由于各自所服务的区域及市场不同,两者在一些细节上存在差异。例如,在溶剂残留标准方面,USP与EP在限度的设定上可能有所不同。又如,某些药物的杂质标准,USP和EP可能会根据不同的科学数据和地区需求做出不同的规定。
然而,这些差异并不妨碍全球药品质量控制的统一性,反而促使药品生产商不断更新其生产和检测工艺,以符合不同市场的要求。
4. 结论
总的来说,USP与EP在药品杂质检测中的标准化作用极为重要。通过为药品生产企业提供统一的检测方法和杂质限度标准,USP与EP帮助确保了全球药品的质量和安全性。无论是在生产、质量控制还是跨国药品注册过程中,遵循USP与EP的标准不仅能够有效控制药品杂质,还能够提升药品的市场竞争力,减少不合规风险。因此,药品生产企业应积极关注并遵循USP与EP在杂质检测中的相关规定,以保障药品质量和患者的健康。

Copyright © 2023-现在 广州佳途科技股份有限公司 版权所有 粤ICP备18108372号-4
CATO标准品信息网为您提供全面标准品、对照品、药物杂质等产品信息,包括品牌、规格、报价等信息,满足实验中标准品/药物杂质采购需求。
地址:广东省广州市黄埔区光谱东路179号百事高智慧园B栋3F-5F 电话:13342851930
地址:广东省广州市黄埔区光谱东路179号百事高智慧园B栋3F-5F
电话:13342851930
企业邮箱:pinfangshu@cato-chem.com 业务QQ:3659756029
业务QQ:3659756029