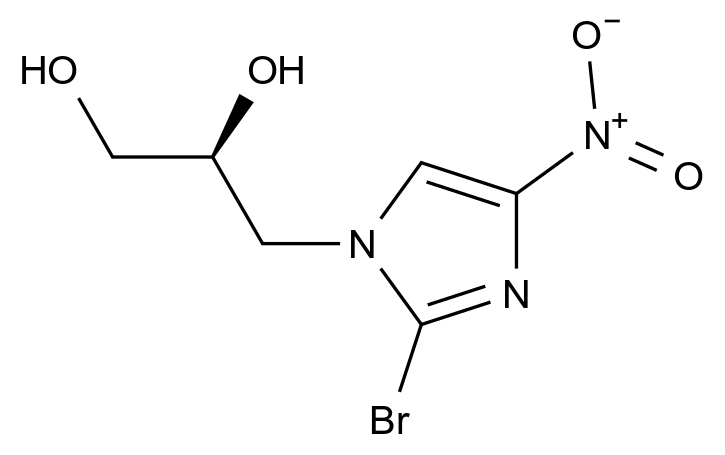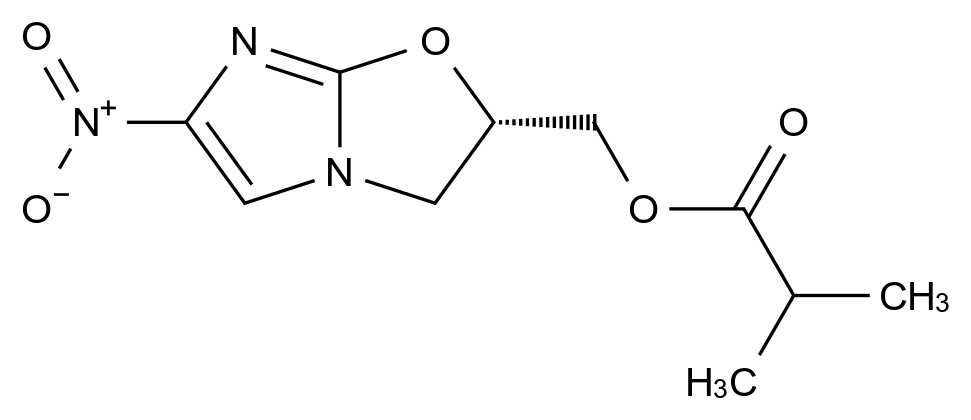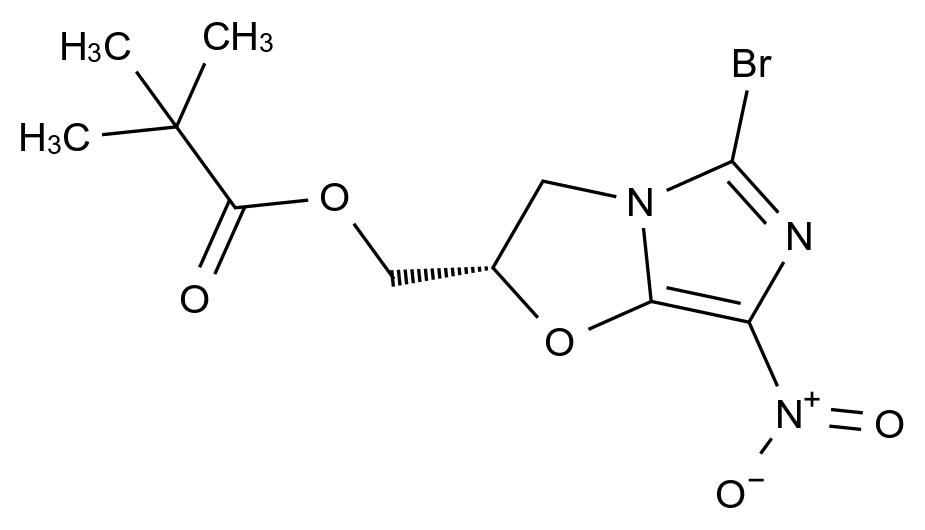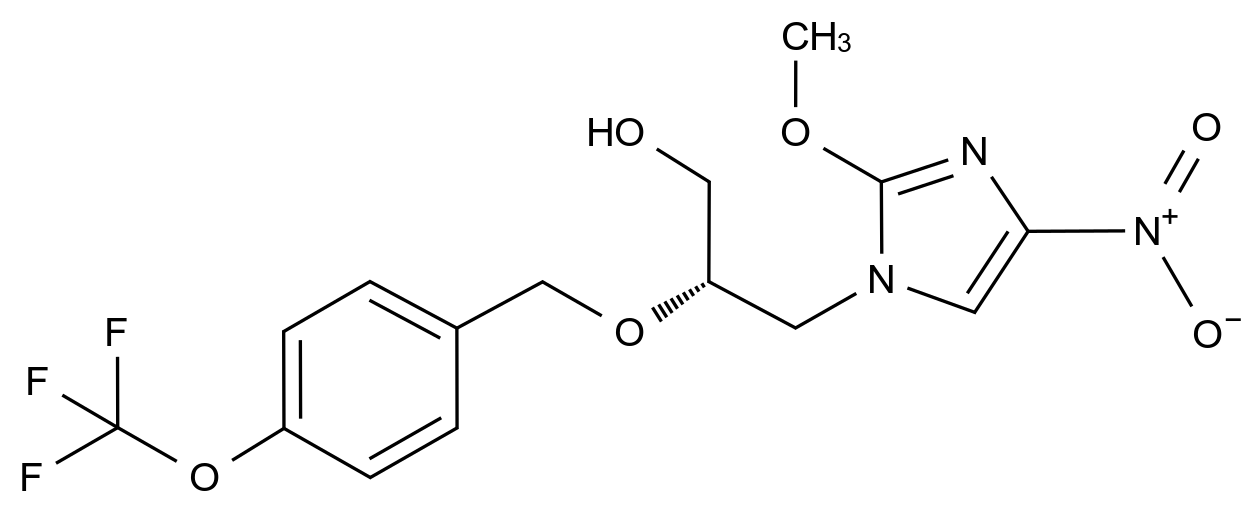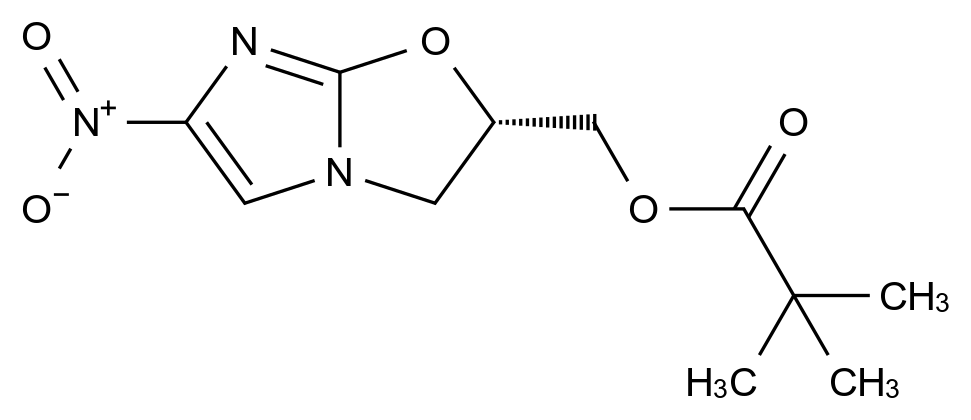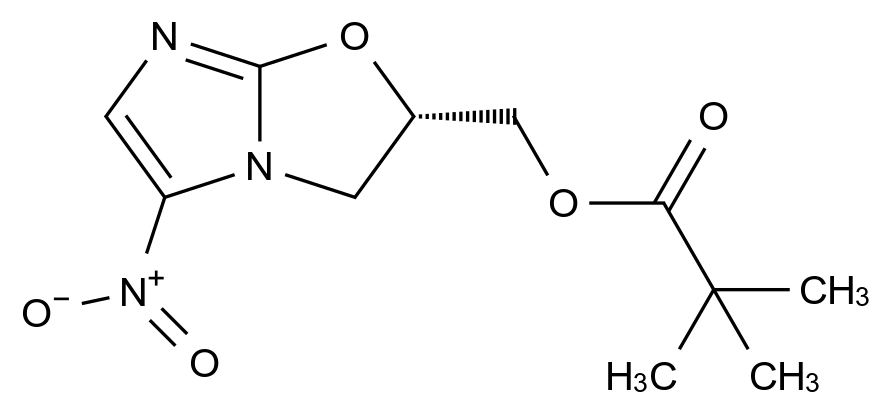
药品稳定性是确保其安全性、有效性和质量一致性的关键因素。随着药品研发的日益精细化和监管要求的不断提高,如何从原料到成品全流程控制杂质、保障稳定性,已成为制药企业核心竞争力的重要体现。
一、稳定性与杂质:相辅相成
药品的“稳定性”指其在储存和使用过程中维持理化性质、纯度和疗效的能力。其核心目标是保证在规定的有效期内,药品不发生降解、变质或产生有害杂质。
而“杂质”包括原料杂质、工艺残留、中间体、副反应产物、降解产物等,它们不仅会影响药品的稳定性,还可能带来安全隐患。例如,部分降解杂质具有潜在毒性,可能引发致敏、致突变甚至致癌风险。
因此,稳定性研究与杂质研究往往是同步推进的,两者在质量控制体系中形成“预防+验证”的闭环结构。
二、从源头管控:原料质量是关键
药品质量的第一道防线在于原料。原料药(API)中若含有杂质、金属离子、水分或残留溶剂,可能在制剂过程中发生进一步反应,导致不良降解。
控制措施包括:
选择稳定性高的晶型或盐型;
优化合成路线,降低反应杂质生成;
使用合格的辅料和包材,避免与原料发生化学反应;
建立完善的原料稳定性档案与供应商审计机制。
三、工艺优化:预防杂质生成
生产工艺的设计直接决定了药品在制造与储存过程中的“应力负荷”。例如:高温干燥、pH剧烈波动、暴露于氧化环境等,都可能加速药品降解。
因此应:
优化制剂处方与工艺参数;
控制水分、氧气、光照等外部条件;
选择合适的包材系统(如PTP铝箔、抗光瓶);
引入质量源于设计(QbD)理念,通过工艺参数与产品性能的系统建模,预判稳定性风险点。
四、稳定性研究:建立科学预测机制
稳定性研究不仅是申报资料的一部分,更是保障产品全生命周期质量的依据。通过加速试验、长期试验、中间条件试验等手段,研究人员可以模拟药品在运输、储存、使用过程中的行为,评估其有效期与储存条件。
同时,应监控关键降解杂质的变化趋势,采用高灵敏度分析方法(如UPLC-MS/MS)进行定量,确保其始终在限度内。
五、监管趋势:杂质谱的透明化
近年来,ICH M7、ICH Q3A/B 等国际指南对基因毒性杂质(GTIs)和未知杂质提出了更为严格的控制要求。EMA、FDA、NMPA等监管机构也日益强调药品“全生命周期杂质追踪”概念。
这意味着:
所有潜在杂质来源必须可追溯;
对未知杂质应建立结构预测与毒理学评估机制;
一旦有新的降解产物被发现,需及时更新稳定性研究方案并补充申报资料。
结语:
药品稳定性不是单一技术点,而是研发、生产、分析、法规、质量控制等多学科协同的结果。只有从源头到终端全链条重视杂质控制与稳定性研究,才能真正实现高质量药品的持续供应,守护每一位患者的用药安全。

Copyright © 2023-现在 广州佳途科技股份有限公司 版权所有 粤ICP备18108372号-4
CATO标准品信息网为您提供全面标准品、对照品、药物杂质等产品信息,包括品牌、规格、报价等信息,满足实验中标准品/药物杂质采购需求。
地址:广东省广州市黄埔区光谱东路179号百事高智慧园B栋3F-5F 电话:13342851930
地址:广东省广州市黄埔区光谱东路179号百事高智慧园B栋3F-5F
电话:13342851930
企业邮箱:pinfangshu@cato-chem.com 业务QQ:3659756029
业务QQ:3659756029