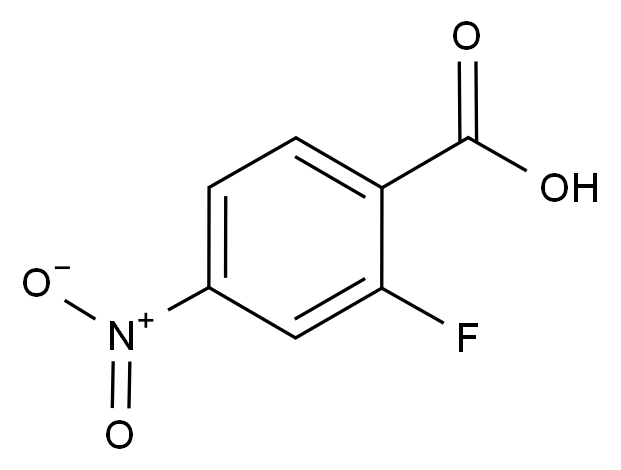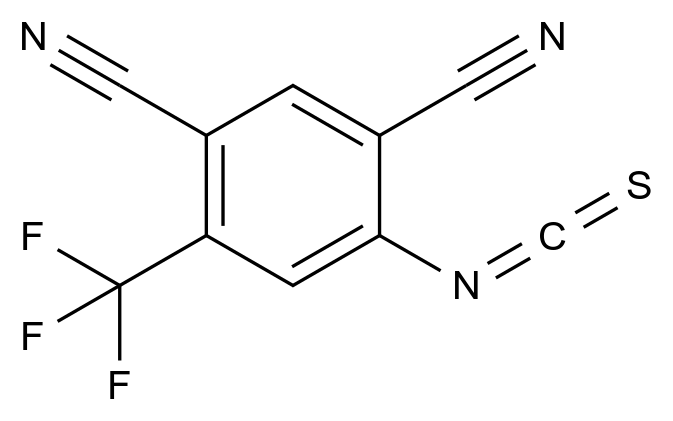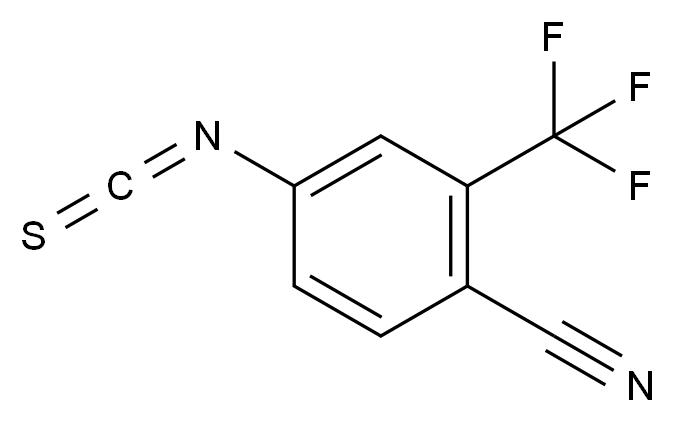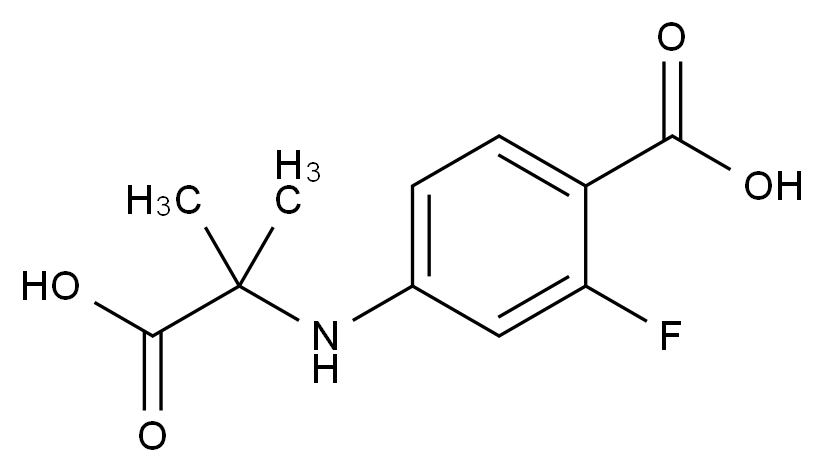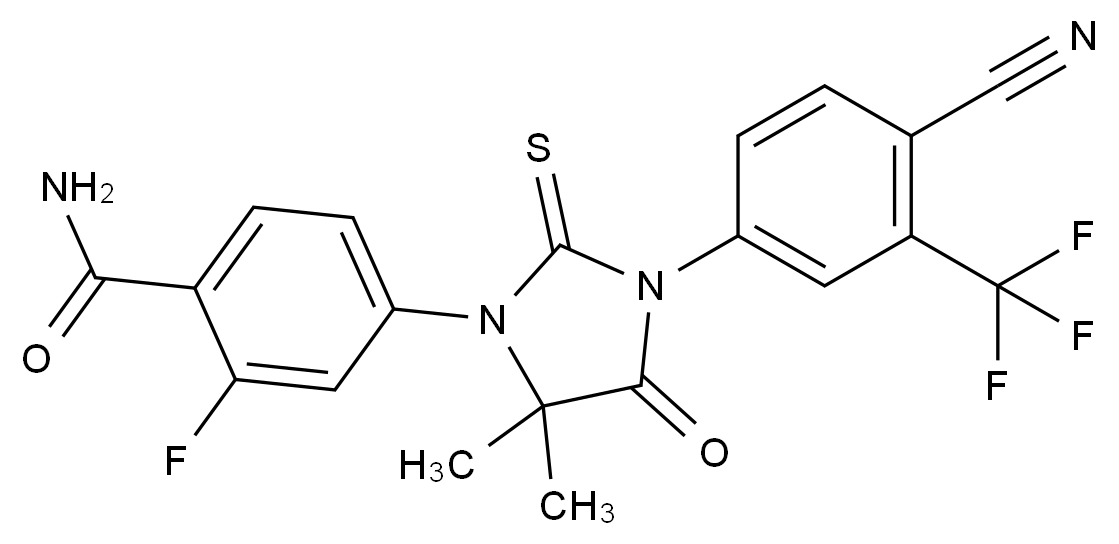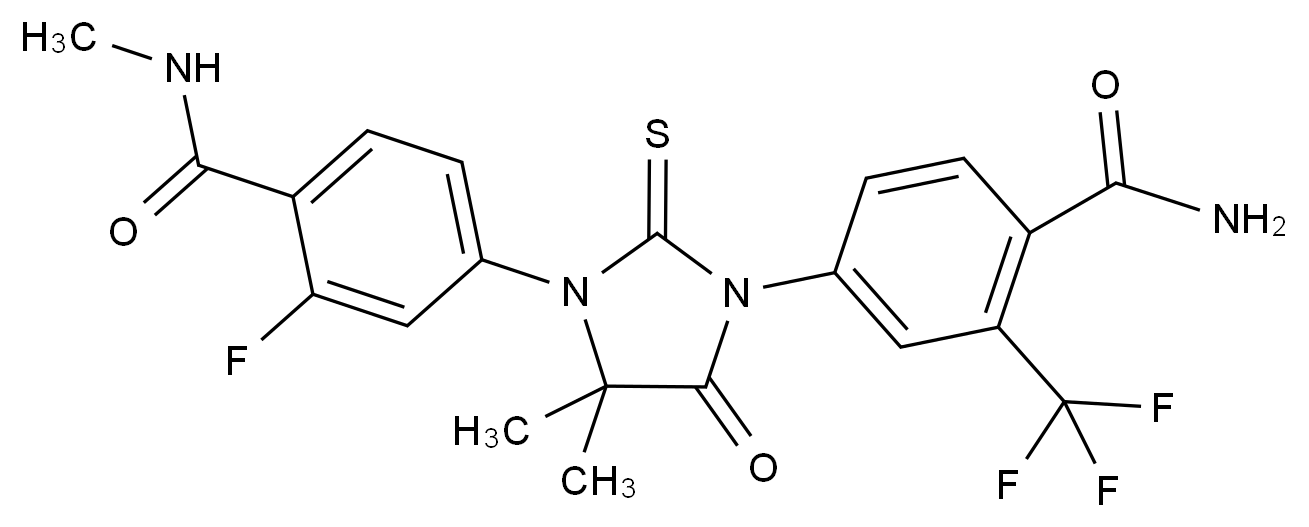
在药品研发与生产过程中,杂质的存在直接关系到产品的安全性、有效性与质量可控性。国际监管机构(如ICH、FDA、EMA)均对杂质限度有严格规范,尤其对基因毒性杂质(GTIs)、工艺杂质、降解产物等提出了明确要求。因此,围绕“减少杂质生成”展开系统性控制,不仅是合规的需求,更是提高产品竞争力的关键。
减少杂质生成的第一步,始于配方阶段的合理设计。药品配方中的每一种成分都可能影响杂质的种类与水平。通过以下方式进行配方优化,可在源头上减少杂质风险:
·
选择稳定性优良的原辅料:避免使用在湿热、光照或氧化条件下易降解的成分,例如可替换易氧化的辅料(如聚山梨酯)为更稳定的替代物。
·
·
避免潜在反应性杂质源:原辅料中可能残留反应性官能团或微量金属离子,这些可在贮存或制备过程中与API发生反应,产生新的杂质。对原辅料进行全面的质量控制与溯源,尤为重要。
·
·
控制pH与溶剂系统:某些杂质的生成对pH高度敏感,如酯类水解、胺类分解反应等。优化处方的pH范围和溶剂极性,能显著降低此类杂质形成。
·
配方确定后,杂质的进一步产生多发生于生产环节。因此,对生产工艺的各个关键参数进行精细控制,是控制杂质的第二道防线。
·
选择适当的反应条件:反应温度、时间、催化剂用量等对杂质生成具有决定性影响。过高的反应温度可能促进副反应,而反应时间不足或过长也可能造成目标产物降解。
杂质机制分析与工艺优化:通过反应动力学与杂质谱研究,识别主杂质来源并回溯其生成路径,结合DOE(Design of Experiment)方法优化反应参数。
·
控制中间体纯度:多个杂质可能源于未完全反应的中间体或副产物,因此需在各中间阶段设置有效的质量控制点(IPC)。
·
优化结晶与萃取过程:部分杂质可能因溶剂选择不当或结晶条件不理想而共晶析出,影响成品纯度。通过精确控制溶剂极性、温度曲线及冷却速率,有助于提高目标产物的纯度。
·
·
设备残留与清洁验证:设备内壁残留物可在批次间迁移,带入未知杂质,需通过定期清洁验证与交叉污染风险评估进行控制。
·
控制微量金属离子与氧含量:某些氧化降解杂质与金属催化反应密切相关,生产中应避免使用可能引入Cu、Fe等金属离子的材料,并尽可能在惰性气氛下操作。
·
减少杂质不仅是生产阶段的任务,也需考虑储存与使用过程中的稳定性。通过加速稳定性试验和长期稳定性试验,可预判贮存条件下可能生成的降解产物。借助高分辨质谱(HRMS)、UPLC-QTOF、NMR等分析手段,对微量未知杂质进行鉴定与定量,进一步完善杂质控制体系。
结语:
从配方设计到工艺控制,再到贮存稳定性监测,减少杂质的生成是一项系统工程。它不仅体现了药品质量控制的“全生命周期”理念,更体现了制药企业对患者用药安全的高度责任感。在监管愈加严格与全球质量标准不断提高的今天,只有将杂质控制“前移”“细化”“量化”,企业才能在合规之上,构建真正的质量优势。

Copyright © 2023-现在 广州佳途科技股份有限公司 版权所有 粤ICP备18108372号-4
CATO标准品信息网为您提供全面标准品、对照品、药物杂质等产品信息,包括品牌、规格、报价等信息,满足实验中标准品/药物杂质采购需求。
地址:广东省广州市黄埔区光谱东路179号百事高智慧园B栋3F-5F 电话:13342851930
地址:广东省广州市黄埔区光谱东路179号百事高智慧园B栋3F-5F
电话:13342851930
企业邮箱:pinfangshu@cato-chem.com 业务QQ:3659756029
业务QQ:3659756029