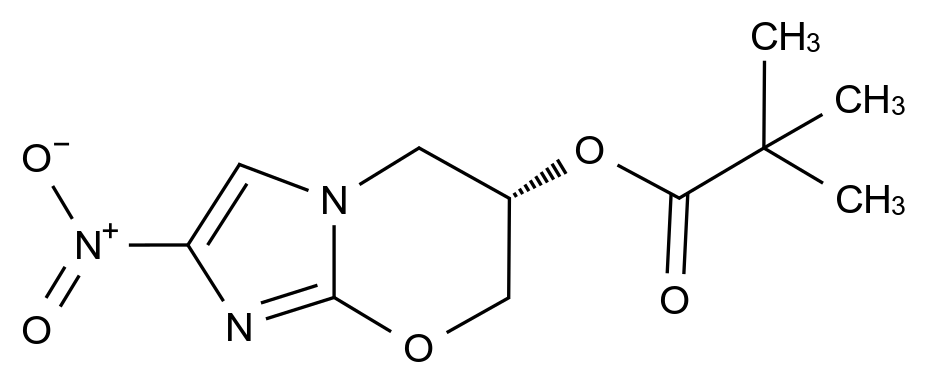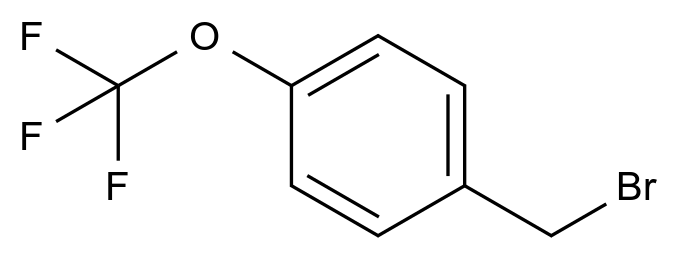
最近,国家药品监督管理局药品审评中心(CDE)连发两份重量级征求意见稿:《化学仿制药生物等效性研究重大缺陷(试行)》与《化学仿制药药学研究重大缺陷(试行)》。这两份文件被业内戏称为仿制药审评的“新断头台”——不是补资料、不是限期整改,而是一旦踩线,即直接退审。

如果说 2017 年的《立卷审查技术标准(暂行)》还只是教大家“资料怎么交齐”“形式怎么合规”,那这两份文件,则直接明确划定了“直接退审”的红线。其中,杂质研究、数据真实性与全过程合规成为了监管层全面围剿的重点,真正从“形式审查”升级到了“实质审评”。
新规之下,药企的“灵魂拷问”
在2017年的立卷标准中,大家关注的是“资料全不全”;而在2025年的新规下,CDE关注的是“研究深不深”。
新规明确指出:“主要质控项目的分析方法不合理”、“长期稳定性考察出现超过鉴定限度的新增杂质且未定性”均属于重大缺陷。 这意味着,如果你的杂质谱没有研究透彻,或者漏掉了关键杂质,项目将面临“直接毙掉”的风险,且没有发补机会。
面对如此严苛的审评环境,药企研发负责人们不禁发出了连串的“灵魂拷问”:
“杂质谱怎么才算研究透了?” 在项目立项初期,我怎么知道这个品种到底有多少潜在杂质?
“去哪找最全的杂质对照品?” 哪一家供应商能提供该品种最完整的杂质列表,让我不至于在审评时被CDE挑战“杂质未定性”?
“质量和资质谁敢保?” 哪一家的CoA数据能经得起CDE对“数据真实性”的显微镜式审查?
“遇到技术难题谁能帮?” 当分析方法需要优化、色谱图处理存在争议时,哪家供应商有能力提供实质性的售后技术支持?”
结合新规,这些困惑可以拆成四个痛点:

新规下CATO给药企提供的解决方案
面对药企的焦虑,围绕“杂质更全、质量更稳、技术更强、响应更快”这四个核心诉求,CATO 凭借深厚的行业积淀,给出了强有力的解决方案。
01 品种覆盖
汇聚全球80%需求,打造“最全杂质库”
CATO 已经深度研究了大量主流仿制品种,覆盖范围可支撑全球约 80% 的市场需求;
对于多数常见及重点品种,CATO 能够提供:
已识别和可合成的杂质清单
与原研/公开文献对照后的杂质映射关系
针对关键/难控杂质,提供专门解决方案(合成路线、分析方法建议等)

这意味着:
在你还处于立项/调研阶段,来找 CATO,就不是“从零开始摸索”,而是站在一个已经验证过的大数据和大样本基础上:
能较早判断:这个品种现在行业里“主流可控的杂质”有哪些、有哪些是难点、哪些是监管重点;可以用更全面的杂质清单来设计自己的质量研究方案,避免后期频繁“被动补课”。
02 质量与体系保障
严苛的质量体系,经得起CDE“溯源”
CATO 已通过 CNAS 与 ANAB 的 ISO 17034 标准物质生产者认证,并按照 ISO/IEC 17025 要求运行检测体系。这意味着我们的产品质量、可追溯性与文件完整性,全部经过国际认可的严格验证。

基于这些体系,CATO 能提供的不仅是“合格的杂质/对照品”,而是一整套“可用于申报”的质量支撑:
批次稳定、文件规范、记录可追溯
完整 CoA、谱图、结构确认与方法学摘要
协助客户在质量标准、方法验证、杂质限度设定上形成完整逻辑链

换句话说,CATO 用完善的质量体系保障把质量不确定性降到最低,让产品经得起审评、经得起追问。
03 技术与售后
专家级技术支持,售后无忧
在当前的监管环境里,“技术支持”不再是锦上添花,而是刚需。
CATO 在技术和售后方面,可以做到:
杂质相关技术咨询:
杂质结构确认、生成路径讨论;分析方法设定建议(如色谱条件、检测波长、梯度等);针对新增杂质、难分离杂质提供可行思路。
配合审评沟通:
如遇到 CDE 针对杂质/对照品提出问题,CATO 能在合理范围内协助提供技术依据或补充解释思路;
售后跟踪:
对重点项目建立持续服务机制,确保在申报关键节点出现问题时,有人能第一时间“接住球”。
04 响应和交付
与仿制药“抢时间”的节奏匹配
新规还规定:“审评期间变更工艺需重新进行稳定性考察”。这意味着时间成本极其昂贵,研发必须争分夺秒。为了配合药企的高效研发,CATO将服务速度做到了极致:
现货报价:10分钟内响应!
非现货报价:24小时内反馈!
配合成熟的供应链和项目管理,使得:
对于常规/热门品种杂质和对照品,能在较短时间内完成发货;
对于定制/特殊杂质,可快速评估可行性、周期和风险,帮助客户尽早做出项目决策。
新规环境下,CATO构筑合规护城河
CDE的两份文件,是一场对医药行业的良币驱逐劣币的阳谋。它清理的是浑水摸鱼者,奖励的是踏实研发者。
在这个容错率极低的时代,您需要的不只是一个供应商,而是一个能帮您把杂质做全、把质量做硬、把响应做快的战略伙伴。
Copyright © 2023-现在 广州佳途科技股份有限公司 版权所有 粤ICP备18108372号-4
CATO标准品信息网为您提供全面标准品、对照品、药物杂质等产品信息,包括品牌、规格、报价等信息,满足实验中标准品/药物杂质采购需求。
地址:广东省广州市黄埔区光谱东路179号百事高智慧园B栋3F-5F 电话:13342851930
地址:广东省广州市黄埔区光谱东路179号百事高智慧园B栋3F-5F
电话:13342851930
企业邮箱:pinfangshu@cato-chem.com 业务QQ:3659756029
业务QQ:3659756029