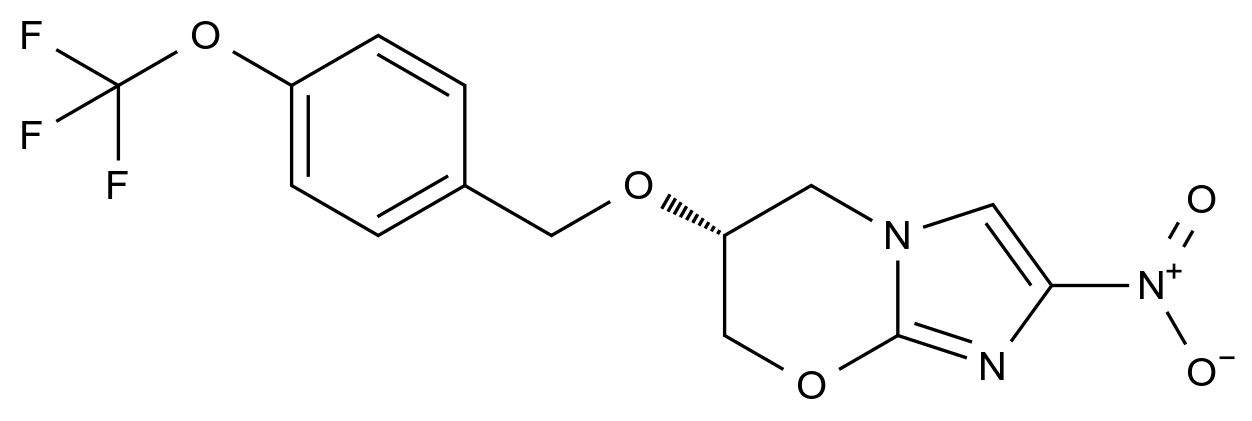
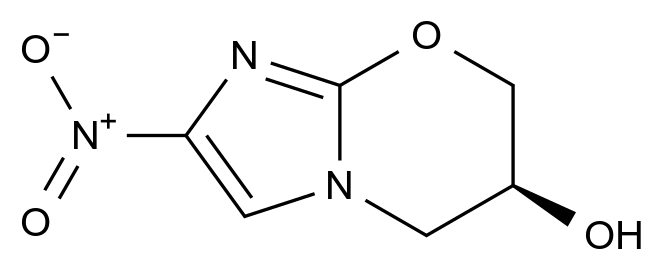
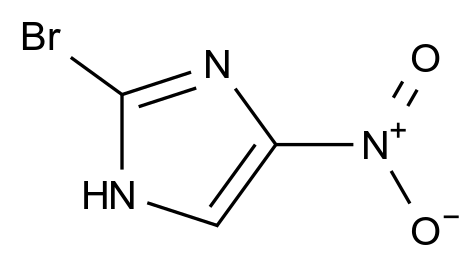
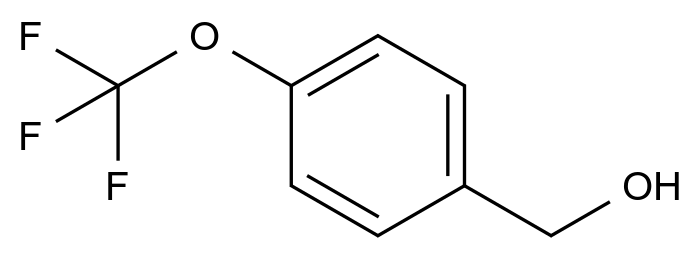
惩前毖后,治病救人。
引言:后专利悬崖时代的创新与仿制新前沿
全球制药行业正处在一个深刻变革的时代。一方面,重磅原研药的专利集中到期引发的“专利悬崖”效应持续发酵,传统小分子仿制药市场因激烈的价格战而利润空间不断被压缩。另一方面,新化学实体(NCE)的研发成本持续攀升,成功率却未见显著提高,迫使整个行业寻求更具成本效益和确定性的增长路径。在这样的宏观背景下,仿制药领域内部正在发生一场深刻的“进化”。这场进化的核心,是告别简单复制的模式,转向技术和知识密集型的高价值领域。
1.解码“复杂仿制药” (Decoding "Complex Generics")
复杂仿制药是仿制药行业向高技术壁垒领域迈进的必然产物。理解其内涵、挑战与机遇,是制定成功研发策略的第一步。
1.1 定义与分类:何为“复杂”?
复杂仿制药并非一个具有严格法律定义的术语,而是一个行业和监管机构普遍接受的概念集合,用以描述那些因其“复杂性”而难以通过传统仿制药路径进行开发和审评的药物。其复杂性主要体现在以下一个或多个维度:
复杂的活性成分 (Complex Active Ingredients):这类药物的活性物质本身就难以完全表征或复制。例如,由多种肽组成的混合物、天然来源的复杂混合物、或者大分子聚合物。它们的分子结构并非单一、明确,给仿制带来了巨大挑战。
复杂的制剂/剂型 (Complex Formulations/Dosage Forms):这是最常见的复杂性来源。原研药可能采用了先进的药物递送系统以实现特定的药代动力学(PK)特性或靶向递送。例如:
脂质体和纳米粒制剂:如多柔比星脂质体,其粒径分布、包封率、表面性质等关键质量属性(CQAs)直接影响药物的体内行为和疗效。
长效缓控释注射剂:通常是基于微球、原位凝胶等技术,可在数周甚至数月内缓慢释放药物,其体外释放(IVR)与体内释放(IVV)的相关性(IVIVC)建立极为困难。
乳剂和混悬剂:如环孢素眼用乳剂,其液滴大小、Zeta电位、晶型等物理化学性质对药物的稳定性和生物利用度至关重要。
复杂的给药途径 (Complex Routes of Delivery):相比于传统的口服或静脉注射,某些给药途径的局部药理作用难以通过全身血药浓度来评估。例如:
局部作用药物:如皮肤科用药膏、吸入剂、滴眼剂等,药物在作用部位(如皮肤、肺部、眼部)的浓度和分布才是关键,而传统的血药浓度BE研究可能不适用。
鼻喷剂和吸入气雾剂:如沙丁胺醇吸入剂,其递送效率、颗粒在呼吸道的沉降部位等都受到制剂处方和吸入装置的双重影响。
复杂的药物-器械组合 (Complex Drug-Device Combinations):这类产品将药物与特定的给药装置(如预充式注射器、自动注射笔、吸入器)相结合。仿制时不仅要复制药物,还必须确保仿制装置的性能与原研装置相当,以保证患者能够获得同等的剂量和使用体验。
在学术和监管语境中,许多复杂仿制药也属于非生物复杂药物(Non-Biological Complex Drugs, NBCDs) 的范畴,这个术语更强调其非生物来源但具有类似生物制品的复杂性。
根据不同的维度,复杂仿制药可以被进一步分类,例如按分子性质(非生物、生物类似药)、给药途径(口服、外用、注射)或治疗领域(肿瘤、自身免疫、神经科学等)进行划分,这有助于企业根据自身技术平台和市场策略进行精准定位。
1.2 核心研发壁垒:技术与法规的双重挑战
攻克复杂仿制药的道路上布满了荆棘,这些障碍既来自科学技术本身,也来自尚在不断演进的法规体系。
技术障碍:等效性证明的“迷雾”
传统仿制药的基石是证明其与原研药的“药学等效性”和“生物等效性”。对于复杂仿制药而言,这一过程变得异常艰难。
生物等效性(BE)评估的困境:对于许多复杂制剂,尤其是局部作用或长效注射剂,经典的、基于健康受试者血药浓度曲线下面积(AUC)和峰浓度(Cmax)的BE研究方法可能不再适用或不足以完全证明治疗等效性(TE)。例如,对于一个治疗皮肤病的药膏,真正重要的是药物在皮肤角质层中的渗透和滞留,而非血液中的浓度。这迫使开发者必须寻找替代的生物等效性评价方法,如药效学(PD)终点研究、体外测试或复杂的临床终点研究,而这些研究往往成本更高、周期更长、不确定性更大。
法规障碍:全球标准不一的“迷宫”
如果说技术障碍是需要攀登的高山,那么法规障碍就像一个路径尚不完全清晰的迷宫。
监管路径与指南的不明确性:尽管近年来全球主要监管机构,特别是美国FDA,在复杂仿制药的审评上取得了长足进步,但许多产品的具体指导原则仍在不断发展和完善中。对于一些全新的复杂剂型,可能根本没有现成的产品特定指南(Product-Specific Guideline, PSG),开发者需要在与监管机构的不断沟通中“摸着石头过河”。
全球监管标准的“碎片化”:不同国家和地区对于复杂仿制药的审评要求存在显著差异。例如,美国FDA通过其505(j)途径和详细的PSG体系,为复杂仿制药提供了一个相对清晰的框架。而欧洲EMA则可能采用“混合申请”(Hybrid Application)途径,并可能要求提供额外的非临床或临床数据来支持等效性声明。对于计划进行全球市场布局的企业而言,这种监管标准的不统一大大增加了研发的复杂度和成本,有时甚至需要为不同市场设计不同的开发策略。
科学与法规的协同挑战:复杂仿制药的审评高度依赖于前沿的监管科学。如何验证新的体外测试方法、如何接受基于模型的模拟数据作为等效性证据,都需要监管机构、学术界和工业界之间进行密切的合作与对话。这种协同机制的建立需要时间,也为个别企业的率先突破带来了不确定性。
2.攻克复杂仿制药的关键策略与前沿技术
面对上述双重挑战,制药行业并未止步不前。在与监管机构的良性互动中,一系列创新的研发策略和前沿技术应运而生,正在逐步铺平通往成功的道路。过去,仿制药企业往往是被动地遵循监管指南。但在复杂仿制药领域,成功的企业必须主动参与到监管科学的建设中,并善用最新的监管工具。
拥抱FDA的领导力与合作倡议:美国FDA在推动复杂仿制药发展方面扮演了全球引领者的角色。其主导的《仿制药使用者付费法案》(GDUFA)为复杂仿制药相关的科学研究提供了大量资金支持,旨在解决关键的科学难题。FDA设立的复杂仿制药研究中心(CRCG)以及频繁举办的研讨会,为行业提供了与监管机构进行早期、深入科学交流的宝贵平台。成功的开发者会积极利用这些机会,通过预申请(Pre-ANDA)会议等机制,在项目早期就关键的科学和技术问题与FDA达成共识,从而有效降低后期研发风险。
范式转变:定量方法与建模(QMM)的应用:面对传统BE研究的局限性,FDA和行业正在大力推动一种新的证据范式——模型整合证据(Model-Integrated Evidence, MIE) 。这是一种将计算机建模与模拟数据与传统的实验数据相结合的综合评估方法。其核心是利用定量方法和模型(QMM)来预测药物在体内的行为,从而为等效性评价提供强有力的支持。
生理药代动力学(PBPK)模型: PBPK模型能够模拟药物在人体内的吸收、分布、代谢和排泄过程。对于复杂口服制剂或注射剂,可以通过构建PBPK模型,论证不同的处方参数(如粒径、溶出速率)对体内PK曲线的影响,从而替代部分人体BE试验。
计算流体动力学(CFD)模型:对于吸入剂和鼻喷剂这类药物-器械组合产品,CFD模型可以精确模拟气流和药物颗粒在呼吸道或鼻腔内的运动和沉降,从而在体外层面证明仿制产品与原研产品在药物递送效率和部位上的等效性。
通过QMM/MIE策略,企业可以在很大程度上减少对昂贵、耗时且存在伦理挑战的人体临床试验的依赖,从而显著加速复杂仿制药的开发进程。
3.探索“超级仿制药”——超越仿制的价值创新
如果说复杂仿制药是在高难度赛道上追求“相同”,那么超级仿制药则是在成熟赛道上创造“不同”,实现价值的跃迁。
声明:以上内容来源网络,如信息有误、需删除或有其他任何疑问,请联系tianwen.zhan@cato-chem.com立刻处理。
Copyright © 2023-现在 广州佳途科技股份有限公司 版权所有 粤ICP备18108372号-4
CATO标准品信息网为您提供全面标准品、对照品、药物杂质等产品信息,包括品牌、规格、报价等信息,满足实验中标准品/药物杂质采购需求。
地址:广东省广州市黄埔区光谱东路179号百事高智慧园B栋3F-5F 电话:13342851930
地址:广东省广州市黄埔区光谱东路179号百事高智慧园B栋3F-5F
电话:13342851930
企业邮箱:pinfangshu@cato-chem.com 业务QQ:3659756029
业务QQ:3659756029